Add external networks to HuMMuS object
Trimbour Rémi
2023-05-16
add_networks.RmdGeneral plan
3. Add a new bipartite
# install python dependency
# reticulate::py_install("hummuspy", envname = "r-reticulate", pip=TRUE)
reticulate::use_condaenv("r-reticulate")
hummuspy <- reticulate::import("hummuspy")
library(HuMMuS)1. Initialisation of HuMMuS object
HuMMuS R objects are instances developed on top of seurat objects. It means it’s created from a seurat object and the contained assays can be accessed the same way.
Additionally, it contains a motifs_db object, providing tf motifs
informations, and a multilayer objects, that will be completed while
going through this tutorial. It will mostly include : - list of
multiplex networks (one per modality) - list of bipartites (one per
connection between layers) 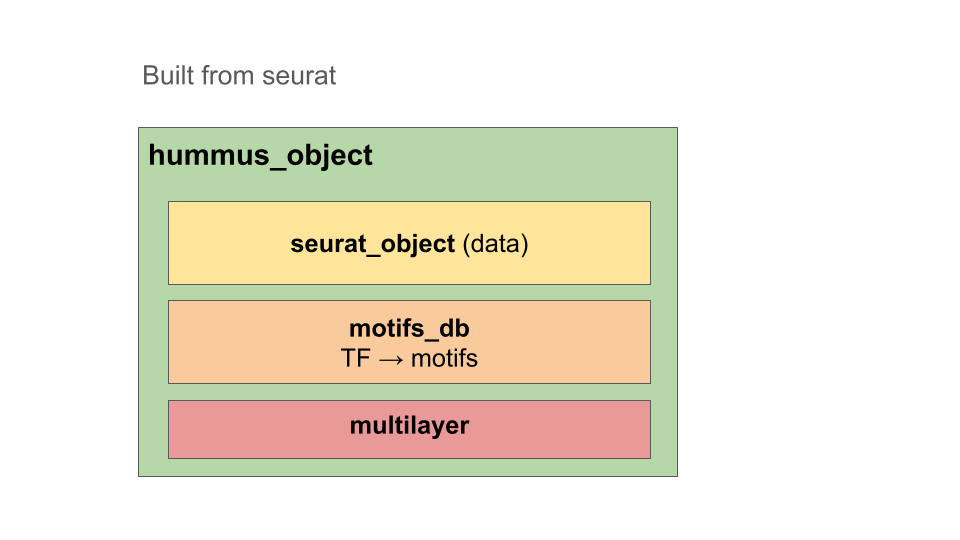
# Create an hummus object from seurat object
# Load the Chen dataset, which is a Seurat object containing scRNA-seq and scATAC-seq data
data("chen_dataset_subset")
chen_dataset_subset## An object of class Seurat
## 12000 features across 385 samples within 2 assays
## Active assay: RNA (2000 features, 0 variable features)
## 2 layers present: counts, data
## 1 other assay present: peaks
hummus <- Initiate_Hummus_Object(chen_dataset_subset)
# wrapper of Signac::GetGRangesFromEnsDb, adapting output to UCSC format
genome_annotations <- get_genome_annotations(
ensdb_annotations = EnsDb.Hsapiens.v86::EnsDb.Hsapiens.v86)
# Add annotations to peak assay
Signac::Annotation(hummus@assays$peaks) <- genome_annotations
rm(genome_annotations)
# Load TF motifs from JASPAR2020 and chromVARmotifs in hummus object
hummus@motifs_db <- get_tf2motifs() # by default human motifs2. Add networks
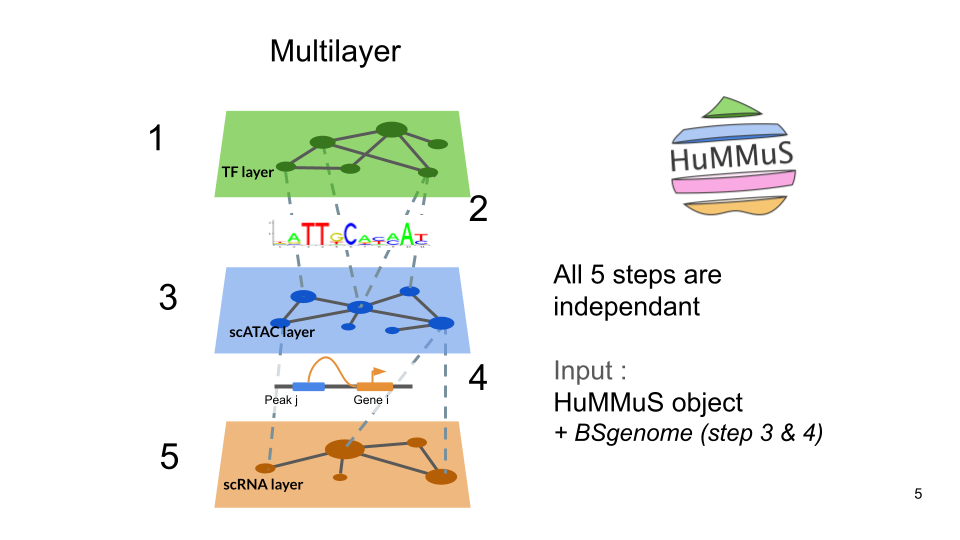
Steps to build HuMMuS objects
2.1 Add a new multiplex through a network
# The networks should be imported as 3 columns data.frame: (source, target, weight)
rna_network_path = "chen_multilayer/multiplex/RNA/RNA_GENIE3.tsv"
rna_network <- read.table(rna_network_path, sep = "\t", header = FALSE)
head(rna_network)## V1 V2 V3
## 1 BACH1 COL4A6 0.7388727
## 2 DDIT3 CCDC173 0.7060954
## 3 DDIT3 CCT6P1 0.6697768
## 4 ATF6B DNASE1 0.6598122
## 5 ARID3B ACER3 0.6499825
## 6 ATF4 ATP8A1 0.6465523
hummus <- add_network(
hummus, rna_network,
multiplex_name = "RNA",
network_name = "GENIE3",
weighted = TRUE,
verbose = 1)## Creating new multiplex : RNA2.2 Add a network to an existing multiplex
hummus <- add_network(
hummus, rna_network,
multiplex_name = "RNA",
network_name = "GENIE3_other",
weighted = TRUE)3 Add external bipartites
Bipartites can be 2 or 3 columns data.frame, depending on if weighted or not. Bipartites not computed through HuMMuS have to be added directly to the object as below:
## Add TF peaks bipartite
atac_rna_path = "chen_multilayer/bipartite/atac_rna.tsv"
atac_rna <- read.table(atac_rna_path, sep = "\t", header = FALSE)
head(atac_rna)## V1 V2
## 1 CMC1 chr3-28241304-28242015
## 2 ANGEL2 chr1-213015141-213016006
## 3 CNNM2 chr10-103053902-103054565
## 4 DNM1L chr12-32678989-32680007
## 5 DHX9 chr1-182838974-182840143
## 6 B3GAT2 chr6-70955759-70956501
hummus@multilayer@bipartites[["atac_rna"]] <- new("bipartite",
"network" = atac_rna,
"multiplex_left" = "RNA",
"multiplex_right" = "peaks")Display HuMMuS object
hummus## Hummus object containing a multilayer object :
## Multilayer network containing 1 bipartite networks and 1 multiplex networks.
##
## - Multiplex names: RNA
## - Bipartite names: atac_rna
##
##
## And a Seurat object :
##
## 12000 features across samples within 2 assays
## Active assay: RNA ( features, 0 variable features)
## 1 other assay present: peaks
hummus@multilayer@multiplex## $RNA
## Multiplex of 2 networks with 2000 features.
## Networks names: GENIE3, GENIE3_other