Predict GRN from scRNA+scATAC data (Chen 2018 dataset)
Trimbour Rémi
2023-05-16
chen_vignette.RmdUseful links:
Paper: https://www.biorxiv.org/content/10.1101/2023.06.09.543828v1
Github repo: https://github.com/cantinilab/HuMMuS
Documentation: https://cantinilab.github.io/HuMMuS/
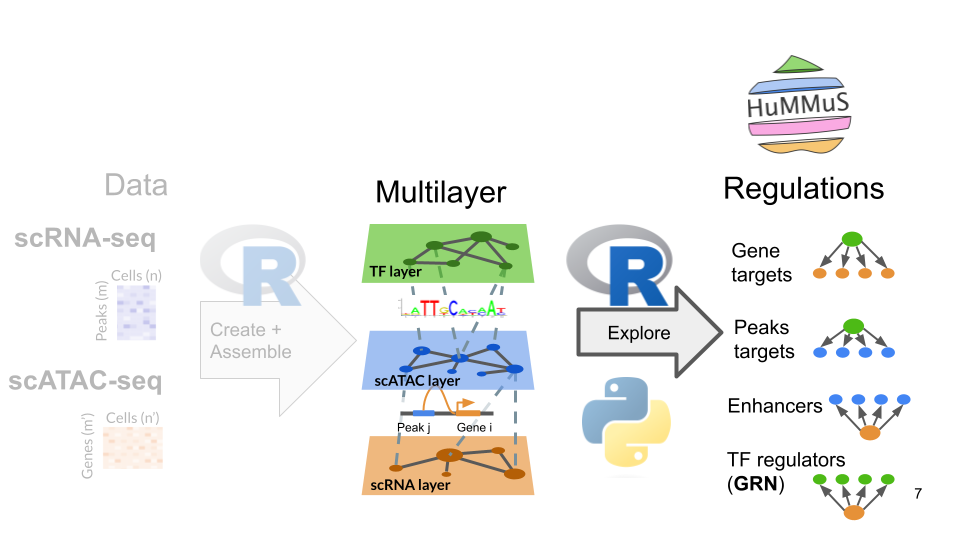
General description of the pipeline
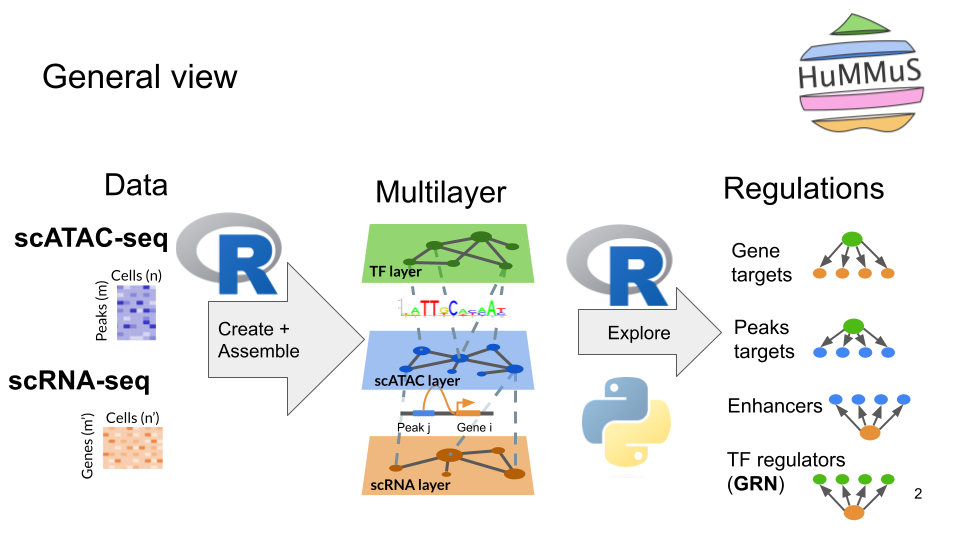
Useful ressources
Preprint detailing the method: https://www.biorxiv.org/content/10.1101/2023.06.09.543828v1
Github repo detailing the installation: https://github.com/cantinilab/HuMMuS
Documentation and vignette: https://cantinilab.github.io/HuMMuS
0. Setting up the environment
# install python dependency
envname = "r-reticulate"
# reticulate::py_install("hummuspy", envname = envname, pip=TRUE)
reticulate::use_condaenv(envname)
hummuspy <- reticulate::import("hummuspy")
library(HuMMuS)Download the single-cell data
The data used in this tutorial can be downloaded here
1. Initialisation of HuMMuS object
HuMMuS R objects are instances developed on top of seurat objects. It means it’s created from a seurat object and the contained assays can be accessed the same way.
Additionally, it contains a motifs_db object, providing tf motifs informations, and a multilayer objects, that will be completed while going through this tutorial. It will mostly include :
- list of multiplex networks (one per modality)
- list of bipartites (one per connection between layers)
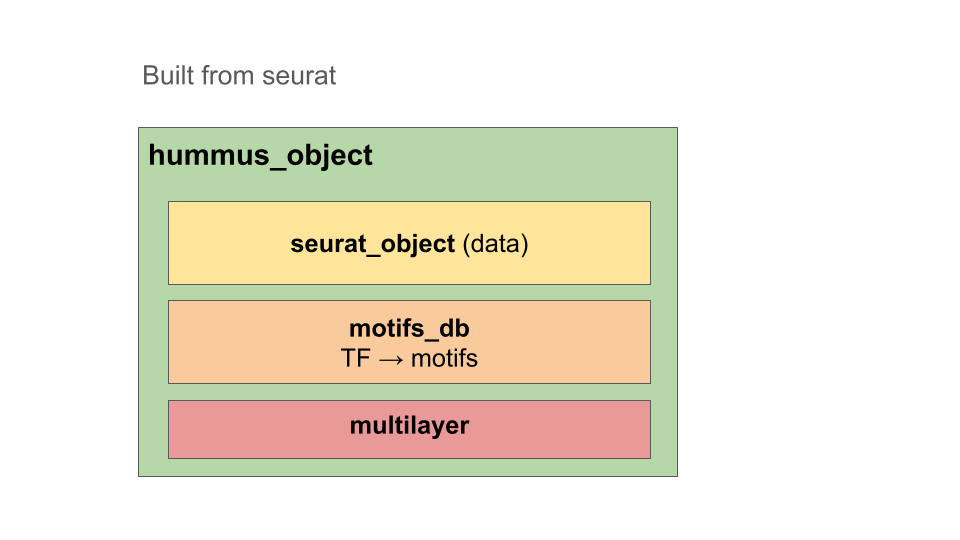
1.1. Transform data into a hummus object
# Load the Chen dataset, which is a Seurat object containing scRNA-seq and scATAC-seq data
data("chen_dataset_subset")
chen_dataset_subset## An object of class Seurat
## 12000 features across 385 samples within 2 assays
## Active assay: RNA (2000 features, 0 variable features)
## 2 layers present: counts, data
## 1 other assay present: peaks
# Create an hummus object from seurat object
hummus <- Initiate_Hummus_Object(chen_dataset_subset)1.2. Add genome and motif annotations to hummus object
Fetch genome annotations online (necessitate an internet connection). You can also request any “EnsDB” object adapted to your data (e.g. EnsDb.Hsapiens.v86::EnsDb.Hsapiens.v86 for human genome annotations) or use your own genome annotations in the same format.
# get human genome annotation from EndDb data
# wrapper of Signac::GetGRangesFromEnsDb, adapting output to UCSC format
genome_annotations <- get_genome_annotations(
ensdb_annotations = EnsDb.Hsapiens.v86::EnsDb.Hsapiens.v86)
# can also be downloaded, saved as an RDS objects for exampleAdd genome annotations to hummus/seurat object
Signac::Annotation(hummus@assays$peaks) <- genome_annotations
rm(genome_annotations)Get TF motifs from JASPAR2020 and chromVARmotifs databsases in a
motifs_db object. By default, human motifs are used. You can specify the
species you want to use with the species argument
(e.g. species = “mouse” for mouse). motifs_db objects contain 3 slots :
* motifs = "PWMatrixList" *
tf2motifs = "data.frame" * tfs = "NULL"
PWMatrixList is a named vector of the motif matrices, whil tf2motifs is
a correspondance table between TFs and motifs. tfs is a named vector of
the TFs. You can also use your own motifs_db object, as long as it
contains the same slots.
# Load TF motifs from JASPAR2020 and chromVARmotifs in hummus object
hummus@motifs_db <- get_tf2motifs() # by default human motifs2. Construction of the multilayer
You can compute the different layers and bipartites as indicated below.
An example multilayer completed can also be imported with :
data(chen_subset_hummus). This object corresponds to a
multilayer from chen_dataset_subset completed. You can then go to the
part 3, replacing hummus by chen_subset_hummus
in each step.
Finally, you can compute the different layers before, and add them afterwards. It allows to use faster methods to compute the networks (e.g. Arboreto for the gene network, ATACNet for the peak network, etc.).
Compute 3 layers and 2 bipartites
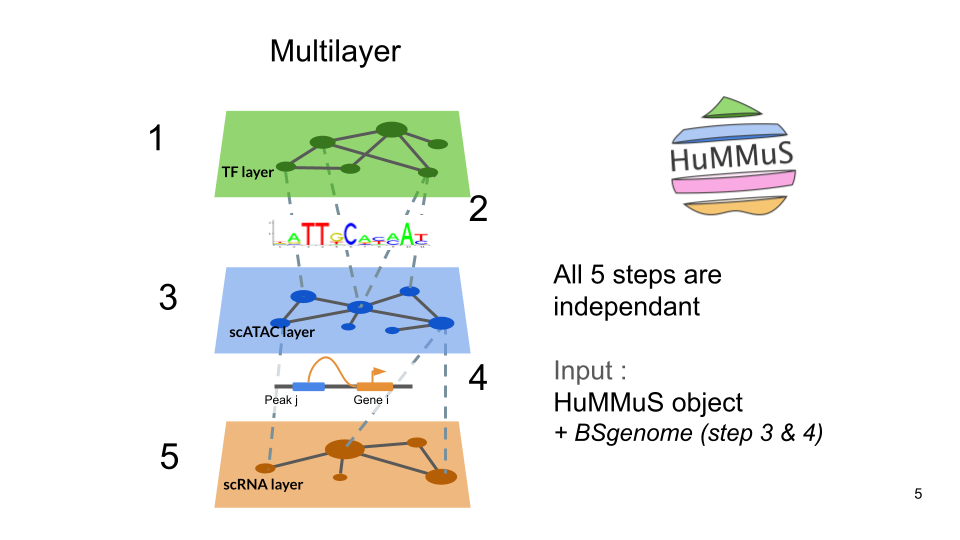
!! Long step !! You can also go directly to the part 3 for your “discovery tour”. :)
2.1. TF - peaks bipartite reconstruction
TF - peaks bipartite is computed using the motifs_db object and the
peak assay. You can specify the assay to use to filter TFs (e.g. “RNA”
if you want to use only the TFs expressed in your dataset). If NULL, all
TFs with motifs will be used. BSGenome object is used to identify
location of motifs and intersect them with peak
You can also specify
the name of the bipartite that will be added to the hummus object. By
default, it will be named “tf_peak”.
hummus <- bipartite_tfs2peaks(
hummus_object = hummus,
tf_expr_assay = "RNA", # use to filter TF on only expressed TFs,
# if NULL, all TFs with motifs are used
peak_assay = "peaks",
tf_multiplex_name = "TF",
genome = BSgenome.Hsapiens.UCSC.hg38::BSgenome.Hsapiens.UCSC.hg38,
)## Computing TF-peak bipartite
## 31 TFs expressed## Building motif matrix## Finding motif positions## Creating Motif object## Warning in CreateMotifObject(data = motif.matrix, positions = motif.positions,
## : Non-unique motif names supplied, making unique## Adding TF info
## Returning TF-peak links as bipartite object
## no peak layer name provided, using peak_assay name2.2. Genes - peaks bipartite reconstruction
Peaks - genes bipartite is computed
hummus <- bipartite_peaks2genes(
hummus_object = hummus,
gene_assay = "RNA",
peak_assay = "peaks",
store_network = FALSE,
)2.3. Compute the TF network from OmniPath database
We currently use OmniPath R package to fetch TF interactions. You can
first specify if you want to use only the TFs expressed in your dataset
(if you have a RNA assay in your hummus object). If
gene_assay is NULL, all TFs with motifs will be used.
You can then specify which interactions you want to keep through
‘source_target’ argument (“AND” | “OR”). If “AND”, only the interactions
between 2 TFs that are both present in the dataset will be kept. If
“OR”, all interactions involving at least one TF present in the dataset
will be kept.
Finally, you can specify the name of the multiplex and
the name of the network that will be added to the hummus object. The
added network will be undirected and unweighted since PPI and OmniPath
database are not directional nor return any weight here.
hummus <- compute_tf_network(hummus,
gene_assay = "RNA", # default = None ;
# If a assay is provided,
# only the TFs that are present
# will be considered
verbose = 1,
#source_target = "OR",
multiplex_name = "TF",
tf_network_name = "TF_network")## Creating a fake TF network with all TFs connected to a fake node.
## 31 TFs expressed
## TF network construction time: 0.007331848 Creating new multiplex : TF!! This step can be very slow if you have thousands of cells !!
Current recommendation if you have a big dataset is to compute
the network before with GRNBoost2 thorugh
arboreto and add
it to the hummus object afterwards. Different methods can be
used to compute the gene network. For now, only GENIE3 is implemented in
HuMMuS. You can specify which assay to use to compute the network
(gene_assay).
You can specify the number of cores to
use to compute the network. You can also specify if you want to save the
network locally (store_network = TRUE) or not
(store_network = FALSE). If you choose to save the network,
you will need to specify the output file name
(output_file). The returned network will be considered
undirected and weighted. While GENIE3 returns a directed network, we
symmetrize it for the random walk with restart exploration of the genes
proximity.
hummus <- compute_gene_network(
hummus,
gene_assay = "RNA",
method = "GENIE3",
verbose = 1,
number_cores = 5, # GENIE3 method can be ran
# parallelised on multiple cores
store_network = FALSE, # by default : FALSE, but
# each network can be saved
# when computed with hummus
output_file = "gene_network.tsv")## Computing gene network with GENIE3 ...
## No TFs list provided, fetching from hummus object...
## 31 TFs expressed
## Gene network construction time: 7.208928
## Creating new multiplex : RNA2.5. Compute the peak network from scATAC-seq w/ Cicero
Different methods can be used to compute the peak network. For now,
only Cicero is implemented in HuMMuS. You can specify which assay to use
to compute the network (peak_assay). You can also specify
the number of cores to use to compute the network. You can also specify
if you want to save the network locally
(store_network = TRUE) or not
(store_network = FALSE). If you choose to save the network,
you will need to specify the output file name
(output_file). The returned network will be considered
undirected and weighted, since cis-regulatory interaction and Cicero
outputs are not directional.
hummus <- compute_atac_peak_network(hummus,
atac_assay = "peaks",
verbose = 1,
genome = BSgenome.Hsapiens.UCSC.hg38::BSgenome.Hsapiens.UCSC.hg38,
store_network = FALSE) ## Overlap QC metrics:
## Cells per bin: 50
## Maximum shared cells bin-bin: 44
## Mean shared cells bin-bin: 6.42882882882883
## Median shared cells bin-bin: 0## Warning in cicero::make_cicero_cds(input_cds, reduced_coordinates =
## umap_coords, : On average, more than 10% of cells are shared between paired
## bins.## [1] "Starting Cicero"
## [1] "Calculating distance_parameter value"## Warning in estimate_distance_parameter(cds, window = window, maxit = 100, :
## Could not calculate sample_num distance_parameters (74 were calculated) - see
## documentation details## [1] "Running models"
## [1] "Assembling connections"
## [1] "Successful cicero models: 4782"
## [1] "Other models: "
##
## Zero or one element in range
## 8803
## [1] "Models with errors: 0"
## [1] "Done"
##
## 4699 peak edges with a coaccess score > 0 were found.
## Peak network construction time: 23.58539 Creating new multiplex : peaks3. Analyse of the multilayer and definition of GRN
data(chen_subset_hummus)
hummus <- chen_subset_hummus3.1. Save the mulilayer in a classic hierarchical structure
The package used for the random walk with restart exploration
(multixrank) requires currently to save all the network files on disk.
To simplify the organisation of the file, it is possible to save
everything necessary with function save_multilayer().
It will create a folder (specified through folder_name)
containing all the files necessary to run the multixrank algorithm. The
folder will contain the following subfolders : *
bipartite : containing the bipartites files *
multiplex : containing the multiplex sub-subfolders *
multiplex_1 (e.g. TF|peak|RNA) : containing the network
file of each layer of the multiplex * seed : that will
contain the seed files (necessary to compute HuMMuS outputs later) *
config : that will contain the config files (necessary
to compute HuMMuS outputs later)
save_multilayer(hummus = hummus,
folder_name = "chen_multilayer")## Warning in dir.create(folder_name): 'chen_multilayer' already exists## Warning in dir.create(paste0(folder_name, "/", multiplex_folder)):
## 'chen_multilayer/multiplex' already exists## Warning in dir.create(paste0(folder_name, "/", bipartite_folder)):
## 'chen_multilayer/bipartite' already exists## Multiplex of 1 networks with 32 features.
## Networks names: TF_network[1] "TF TF_network"## Warning in dir.create(paste0(folder_name, "/", multiplex_folder, "/",
## multiplex_name)): 'chen_multilayer/multiplex/RNA' already exists## Multiplex of 1 networks with 2000 features.
## Networks names: RNA_GENIE3[1] "RNA RNA_GENIE3"
## Multiplex of 1 networks with 4639 features.
## Networks names: peak_network_cicero[1] "peaks peak_network_cicero"3.2. Retrieve target genes
With HuMMuS, inference of GRN and target gene of TFs are different
outputs. Indeed, while GRN is computed making TFs compete to regulate
genes (by random walk with restart starting from the genes and going to
the TFs), target genes are computed making genes compete to be regulated
by TFs (by random walk with restart starting from the TFs and going to
the genes).
For target genes output, you can specify the list of TFs
(tf_list) to use as seed (if NULL by default, all TFs will
be used as seed). Only the links between the seed TFs and the genes will
be computed. You can also specify the list of genes to use. Only the
score of the genes present in the network and the gene_list
will be returned.
ATF2_genes <- define_target_genes(
hummus,
tf_list = list("ATF2"),
multilayer_f = "chen_multilayer",
njobs = 1
)## multiplexes_list : {'TF': {'TF_network': '00'}, 'RNA': {'RNA_GENIE3': '01'}, 'peaks': {'peak_network_cicero': '01'}}
## bipartites_list : {'tf_peak.tsv': {'multiplex_right': 'TF', 'multiplex_left': 'peaks'}, 'atac_rna.tsv': {'multiplex_right': 'peaks', 'multiplex_left': 'RNA'}}
## folder_multiplexes : multiplex
## folder_bipartites : bipartite
## gene_list : None
## tf_list : ['ATF2']
## peak_list : None
## config_filename : target_genes_config.yml
## config_folder : config
## tf_multiplex : TF
## peak_multiplex : peaks
## rna_multiplex : RNA
## update_config : True
## save : False
## return_df : True
## output_f : None
## njobs : 1
## {'TF': {'TF_network': '00'}, 'RNA': {'RNA_GENIE3': '01'}, 'peaks': {'peak_network_cicero': '01'}}
## bipartites_type has been provided throguh a list, make sure the order matches the one of the 'bipartites' dictionary' keys.
## RNA_1
## Opening network from chen_multilayer/multiplex/RNA/RNA_GENIE3.tsv.
## TF_1
## Opening network from chen_multilayer/multiplex/TF/TF_network.tsv.
## peaks_1
## Opening network from chen_multilayer/multiplex/peaks/peak_network_cicero.tsv.
## Opening network from chen_multilayer/bipartite/tf_peak.tsv.
## Opening network from chen_multilayer/bipartite/atac_rna.tsv.
## http://127.0.0.1:8787/status
## Processing seeds... ━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 100% 0:00:00
head(ATF2_genes)## index layer path_layer score tf gene
## 1 1175 RNA RNA_1 5.291440e-05 ATF2 CDC5L
## 2 540 RNA RNA_1 4.570328e-05 ATF2 ATF3
## 3 591 RNA RNA_1 4.486208e-05 ATF2 ATP5I
## 4 1096 RNA RNA_1 4.485191e-05 ATF2 CCNB1IP1
## 5 1872 RNA RNA_1 4.482241e-05 ATF2 DGCR14
## 6 536 RNA RNA_1 4.176609e-05 ATF2 ATAT1
target_genes <- define_target_genes(
hummus,
multilayer_f = "chen_multilayer",
njobs = 1
)## multiplexes_list : {'TF': {'TF_network': '00'}, 'RNA': {'RNA_GENIE3': '01'}, 'peaks': {'peak_network_cicero': '01'}}
## bipartites_list : {'tf_peak.tsv': {'multiplex_right': 'TF', 'multiplex_left': 'peaks'}, 'atac_rna.tsv': {'multiplex_right': 'peaks', 'multiplex_left': 'RNA'}}
## folder_multiplexes : multiplex
## folder_bipartites : bipartite
## gene_list : None
## tf_list : None
## peak_list : None
## config_filename : target_genes_config.yml
## config_folder : config
## tf_multiplex : TF
## peak_multiplex : peaks
## rna_multiplex : RNA
## update_config : True
## save : False
## return_df : True
## output_f : None
## njobs : 1
## {'TF': {'TF_network': '00'}, 'RNA': {'RNA_GENIE3': '01'}, 'peaks': {'peak_network_cicero': '01'}}
## bipartites_type has been provided throguh a list, make sure the order matches the one of the 'bipartites' dictionary' keys.
## RNA_1
## Opening network from chen_multilayer/multiplex/RNA/RNA_GENIE3.tsv.
## TF_1
## Opening network from chen_multilayer/multiplex/TF/TF_network.tsv.
## peaks_1
## Opening network from chen_multilayer/multiplex/peaks/peak_network_cicero.tsv.
## Opening network from chen_multilayer/bipartite/tf_peak.tsv.
## Opening network from chen_multilayer/bipartite/atac_rna.tsv.
## http://127.0.0.1:8787/status
## Processing seeds... ━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 100% 0:00:02
head(target_genes)## index layer path_layer score tf gene
## 1 1340 RNA RNA_1 0.0002318541 CUX1 CHRNB1
## 2 1175 RNA RNA_1 0.0002299783 CUX1 CDC5L
## 3 262 RNA RNA_1 0.0002217501 CUX1 ALG11
## 4 1182 RNA RNA_1 0.0002211187 CUX1 CDCA5
## 5 196 RNA RNA_1 0.0002201921 CUX1 AGBL5
## 6 1172 RNA RNA_1 0.0001498125 BPTF CDC42SE13.3. Define GRN
The GRN is defined using the multixrank algorithm. It requires to
have the hummuspy python package installed (pip install hummuspy).
This can be parallelised using the njobs argument. You can also
specify the list of genes and the list of TFs to use.
grn <- define_grn(
hummus,
multilayer_f = "chen_multilayer",
njobs = 5
)
grn3.4. Retrieve enhancers
You can also specify the list of peaks to use.
enhancers <- define_enhancers(
hummus,
gene_list = list("ATF2"),
multilayer_f = "chen_multilayer",
njobs = 1
)## multiplexes_list : {'TF': {'TF_network': '00'}, 'RNA': {'RNA_GENIE3': '01'}, 'peaks': {'peak_network_cicero': '01'}}
## bipartites_list : {'tf_peak.tsv': {'multiplex_right': 'TF', 'multiplex_left': 'peaks'}, 'atac_rna.tsv': {'multiplex_right': 'peaks', 'multiplex_left': 'RNA'}}
## folder_multiplexes : multiplex
## folder_bipartites : bipartite
## gene_list : ['ATF2']
## tf_list : None
## peak_list : None
## config_filename : enhancers_config.yml
## config_folder : config
## tf_multiplex : TF
## peak_multiplex : peaks
## rna_multiplex : RNA
## update_config : True
## save : False
## return_df : True
## output_f : None
## njobs : 1
## {'TF': {'TF_network': '00'}, 'RNA': {'RNA_GENIE3': '01'}, 'peaks': {'peak_network_cicero': '01'}}
## bipartites_type has been provided throguh a list, make sure the order matches the one of the 'bipartites' dictionary' keys.
## RNA_1
## Opening network from chen_multilayer/multiplex/RNA/RNA_GENIE3.tsv.
## TF_1
## Opening network from chen_multilayer/multiplex/TF/TF_network.tsv.
## peaks_1
## Opening network from chen_multilayer/multiplex/peaks/peak_network_cicero.tsv.
## Opening network from chen_multilayer/bipartite/tf_peak.tsv.
## Opening network from chen_multilayer/bipartite/atac_rna.tsv.
## http://127.0.0.1:8787/status
## Processing seeds... ━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 100% 0:00:00
head(enhancers)## index layer path_layer score gene peak
## 1 3950 peaks peaks_1 0.0008951819 ATF2 chr6-44387360-44388305
## 2 3134 peaks peaks_1 0.0007383304 ATF2 chr3-107522766-107524070
## 3 4007 peaks peaks_1 0.0006763254 ATF2 chr7-102125135-102125606
## 4 4189 peaks peaks_1 0.0004115710 ATF2 chr7-73738654-73739271
## 5 676 peaks peaks_1 0.0003972408 ATF2 chr10-73874242-73875104
## 6 1699 peaks peaks_1 0.0003850920 ATF2 chr16-67561885-675635103.5. Retrieve binding regions
For binding regions output, you can specify the list of TFs
(tf_list) to use as seed (if NULL by default, all TFs will
be used as seed). Only the links between the seed TFs and the peaks will
be computed. You can also specify the list of peaks to use. Only the
score of the peaks present in the network and the peak_list
will be returned.
binding_regions <- define_binding_regions(
hummus,
multilayer_f = "chen_multilayer",
njobs = 1
)## multiplexes_list : {'TF': {'TF_network': '00'}, 'RNA': {'RNA_GENIE3': '01'}, 'peaks': {'peak_network_cicero': '01'}}
## bipartites_list : {'tf_peak.tsv': {'multiplex_right': 'TF', 'multiplex_left': 'peaks'}, 'atac_rna.tsv': {'multiplex_right': 'peaks', 'multiplex_left': 'RNA'}}
## folder_multiplexes : multiplex
## folder_bipartites : bipartite
## gene_list : None
## tf_list : None
## peak_list : None
## config_filename : binding_regions_config.yml
## config_folder : config
## tf_multiplex : TF
## peak_multiplex : peaks
## rna_multiplex : RNA
## update_config : True
## save : False
## return_df : True
## output_f : None
## njobs : 1
## {'TF': {'TF_network': '00'}, 'RNA': {'RNA_GENIE3': '01'}, 'peaks': {'peak_network_cicero': '01'}}
## bipartites_type has been provided throguh a list, make sure the order matches the one of the 'bipartites' dictionary' keys.
## RNA_1
## Opening network from chen_multilayer/multiplex/RNA/RNA_GENIE3.tsv.
## TF_1
## Opening network from chen_multilayer/multiplex/TF/TF_network.tsv.
## peaks_1
## Opening network from chen_multilayer/multiplex/peaks/peak_network_cicero.tsv.
## Opening network from chen_multilayer/bipartite/tf_peak.tsv.
## Opening network from chen_multilayer/bipartite/atac_rna.tsv.
## http://127.0.0.1:8787/status
## Processing seeds... ━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 100% 0:00:02
head(binding_regions)## index layer path_layer score tf peak
## 1 464 peaks peaks_1 0.001680402 CUX1 chr1-65002512-65003322
## 2 4617 peaks peaks_1 0.001675393 CUX1 chrX-48971714-48972653
## 3 468 peaks peaks_1 0.001674121 CUX1 chr1-65419692-65420979
## 4 4615 peaks peaks_1 0.001559648 CUX1 chrX-48918888-48919598
## 5 4307 peaks peaks_1 0.001502478 CUX1 chr8-24955892-24956818
## 6 2655 peaks peaks_1 0.001502093 CUX1 chr2-201642365-201643651